one hot encoding for CDR3
encode_one_cdr3<- function(cdr3, length_cutoff){
res<- matrix(0, nrow = length(total_aa), ncol = length_cutoff)
cdr3<- unlist(stringr::str_split(cdr3, pattern = ""))
cdr3<- cdr3[1:length_cutoff]
row_index<- sapply(cdr3, function(x) which(total_aa==x)[1])
col_index<- 1: length_cutoff
res[as.matrix(cbind(row_index, col_index))]<- 1
return(res)
}
cdr3<- "CASSLKPNTEAFF"
encode_one_cdr3(cdr3, length_cutoff = 12)#> [,1] [,2] [,3] [,4] [,5] [,6] [,7] [,8] [,9] [,10] [,11] [,12]
#> [1,] 0 1 0 0 0 0 0 0 0 0 1 0
#> [2,] 0 0 0 0 0 0 0 0 0 0 0 0
#> [3,] 0 0 0 0 0 0 0 1 0 0 0 0
#> [4,] 0 0 0 0 0 0 0 0 0 0 0 0
#> [5,] 1 0 0 0 0 0 0 0 0 0 0 0
#> [6,] 0 0 0 0 0 0 0 0 0 1 0 0
#> [7,] 0 0 0 0 0 0 0 0 0 0 0 0
#> [8,] 0 0 0 0 0 0 0 0 0 0 0 0
#> [9,] 0 0 0 0 0 0 0 0 0 0 0 0
#> [10,] 0 0 0 0 0 0 0 0 0 0 0 0
#> [11,] 0 0 0 0 1 0 0 0 0 0 0 0
#> [12,] 0 0 0 0 0 1 0 0 0 0 0 0
#> [13,] 0 0 0 0 0 0 0 0 0 0 0 0
#> [14,] 0 0 0 0 0 0 0 0 0 0 0 1
#> [15,] 0 0 0 0 0 0 1 0 0 0 0 0
#> [16,] 0 0 1 1 0 0 0 0 0 0 0 0
#> [17,] 0 0 0 0 0 0 0 0 1 0 0 0
#> [18,] 0 0 0 0 0 0 0 0 0 0 0 0
#> [19,] 0 0 0 0 0 0 0 0 0 0 0 0
#> [20,] 0 0 0 0 0 0 0 0 0 0 0 0This gives you a 20 x 12 matrix, the entry is 1 when the aa is matching the total_aa.
e.g., the first aa is C, so the [5, 1] is 1.
shape the training and testing data
Read my previous post on tensor reshaping.
length_cutoff = 12
train_data<- purrr::map(train_CDR3$X1, ~encode_one_cdr3(.x, length_cutoff = length_cutoff))
# reshape the data to a 2D tensor
train_data<- array_reshape(unlist(train_data), dim = c(length(train_data), 20 * length_cutoff))
# the 20x12 matrix is linearized to a 240 element vector
dim(train_data)#> [1] 70000 240test_data<- purrr::map(test_CDR3$X1, ~encode_one_cdr3(.x, length_cutoff = length_cutoff))
test_data<- array_reshape(unlist(test_data), dim= c(length(test_data), 20* length_cutoff))
dim(test_data)#> [1] 29851 240The original paper used a Convolutional Neural Network (CNN), I will use a regular 2 dense-layer model.
y_train <- as.numeric(train_label)
y_test <- as.numeric(test_label)
model <- keras_model_sequential() %>%
layer_dense(units = 16, activation = "relu", input_shape = c(20 * length_cutoff)) %>%
layer_dense(units = 16, activation = "relu") %>%
layer_dense(units = 1, activation = "sigmoid")
model %>% compile(
optimizer = "rmsprop",
loss = "binary_crossentropy",
metrics = c("accuracy")
)If one use big epochs, the model will be over-fitted. Set apart 35000 CDR3 sequences for validation.
set.seed(123)
val_indices <- sample(nrow(train_data), 35000)
x_val <- train_data[val_indices,]
partial_x_train <- train_data[-val_indices,]
y_val <- y_train[val_indices]
partial_y_train <- y_train[-val_indices]
history <- model %>% fit(partial_x_train,
partial_y_train,
epochs = 20,
batch_size = 512,
validation_data = list(x_val, y_val)
)
plot(history)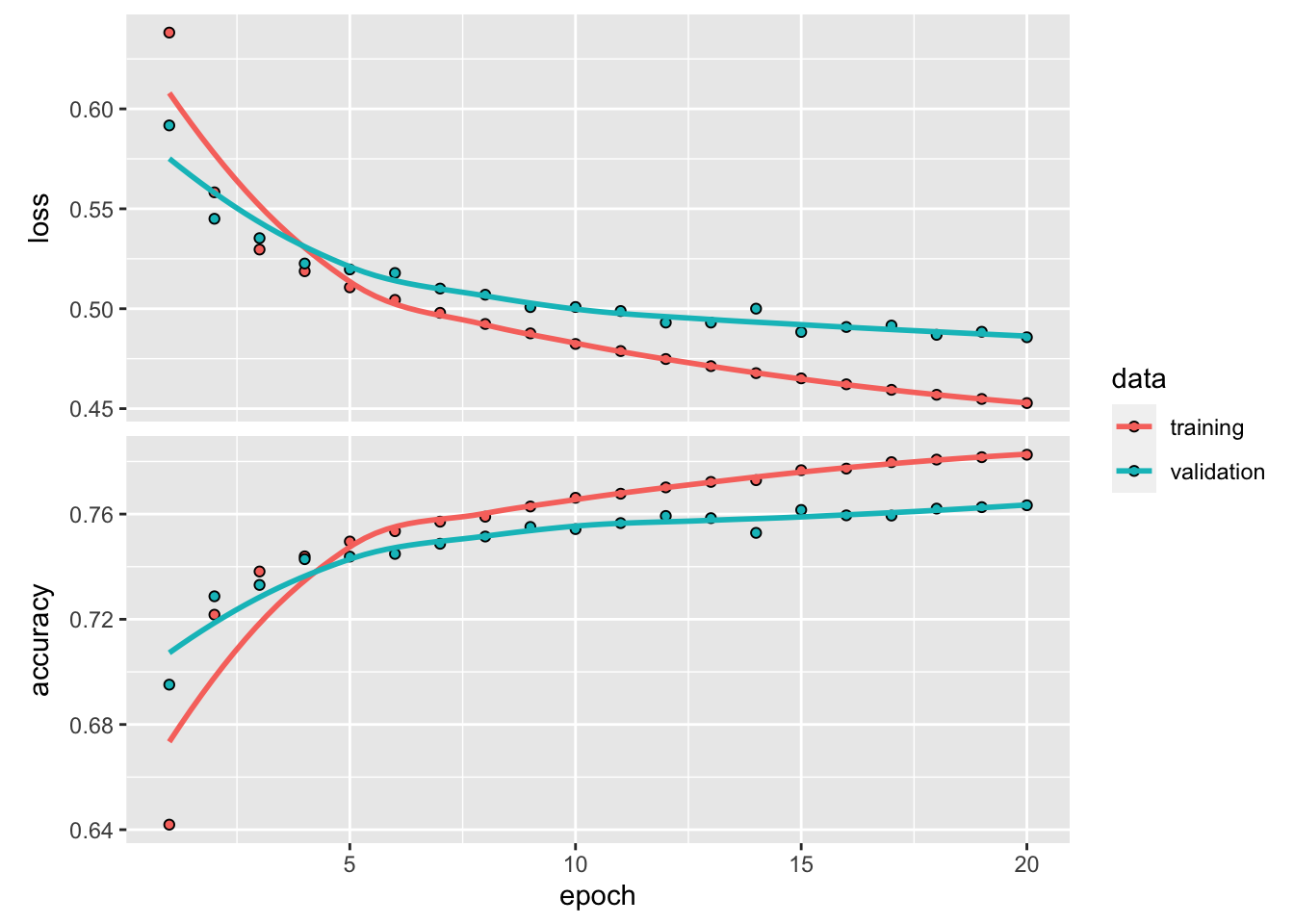
final training and testing
model %>% fit(train_data, y_train, epochs = 20, batch_size = 512)
results <- model %>% evaluate(test_data, y_test)
results#> loss accuracy
#> 0.4229931 0.8065391~80% accuracy. Not bad…
Let’s increase the number of units in each layer.
model <- keras_model_sequential() %>%
layer_dense(units = 64, activation = "relu", input_shape = c(20 * length_cutoff)) %>%
layer_dense(units = 64, activation = "relu") %>%
layer_dense(units = 1, activation = "sigmoid")
model %>% compile(
optimizer = "rmsprop",
loss = "binary_crossentropy",
metrics = c("accuracy")
)
val_indices <- sample(nrow(train_data), 35000)
x_val <- train_data[val_indices,]
partial_x_train <- train_data[-val_indices,]
y_val <- y_train[val_indices]
partial_y_train <- y_train[-val_indices]
history <- model %>% fit(partial_x_train,
partial_y_train,
epochs = 20,
batch_size = 512,
validation_data = list(x_val, y_val)
)
plot(history)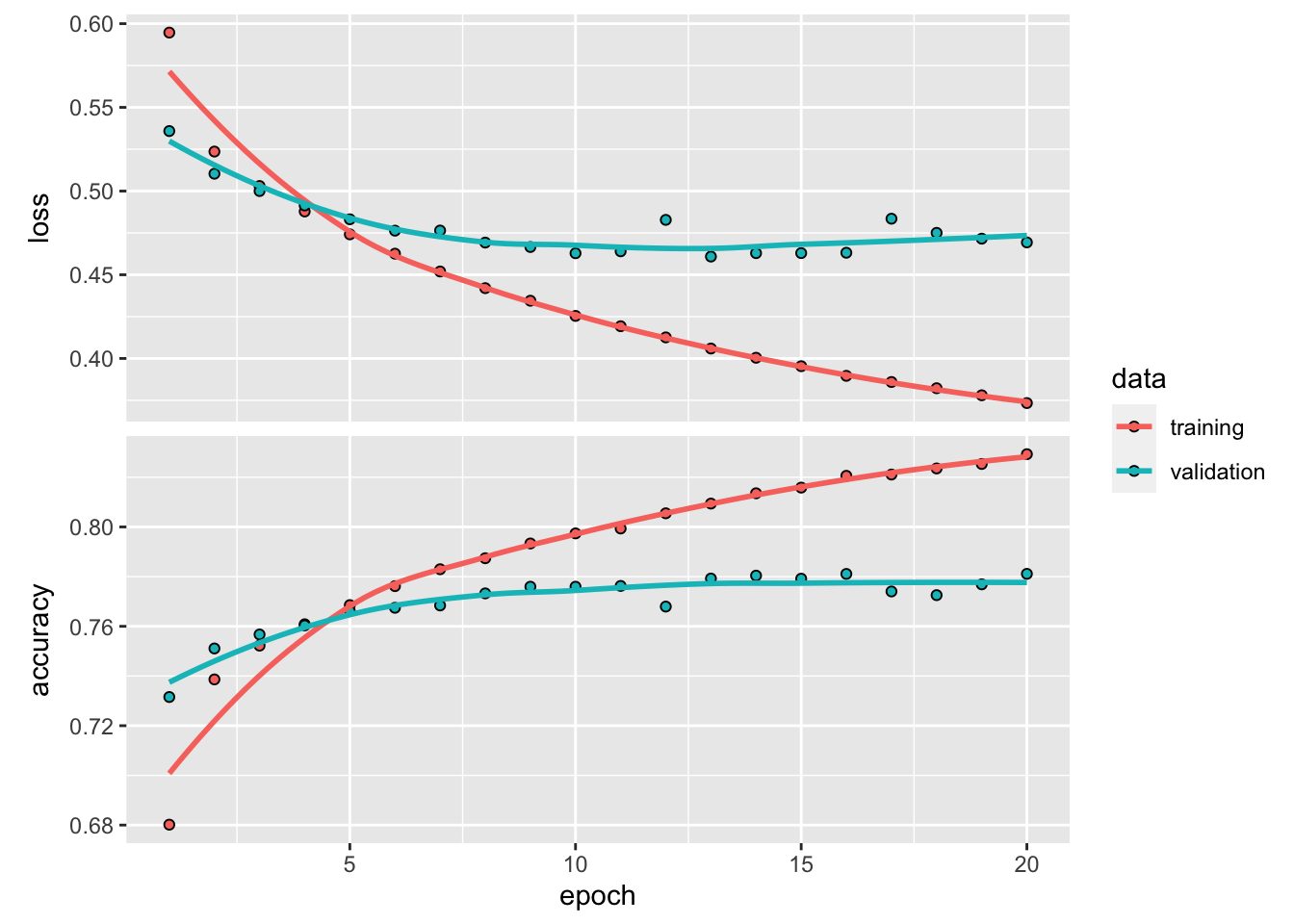
with more units (neurons) in each layer, overfitting occurs much faster. after 10 epoch, the validation accuracy starts to plateau.
model %>% fit(train_data, y_train, epochs = 10, batch_size = 512)
results <- model %>% evaluate(test_data, y_test)
results#> loss accuracy
#> 0.3731814 0.8282135predict(model, test_data[1:10, ])#> [,1]
#> [1,] 0.08662453
#> [2,] 0.23022524
#> [3,] 0.21670932
#> [4,] 0.03327328
#> [5,] 0.03596783
#> [6,] 0.07136077
#> [7,] 0.06769678
#> [8,] 0.03962836
#> [9,] 0.16300359
#> [10,] 0.36324140How to improve the accuracy by using biology domian knowledge?
It is surprising to me that using only the CDR3 aa sequences can reach an accuracy of 80%. How can we further improve it?
- we can add the AA properties
- add the VDJ usage
- add HLA typing for each sample