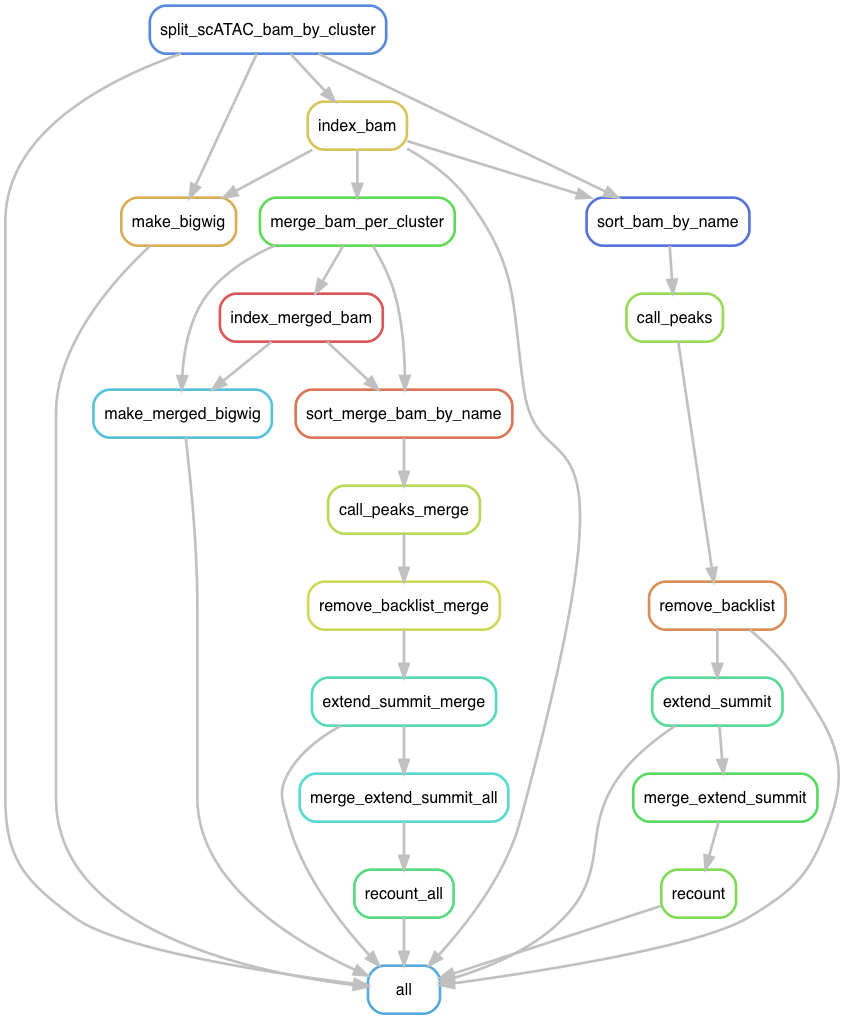
What does it do?
For single cell ATACseq experiment, one gets a merged bam file for all cells. After going through clustering, one groups similar cells into cell types (cell states). This workflow will split the bam by clusters to create a pseudo bulk bam for each cluster, create bigwig tracks for visulization, call peaks for each cluster and merge the peaks across the clusters. Finally it will count reads per peak per cell from the original bam file on the merged peaks.
In the future, the peak calling software should be barcode aware, so one does not need to split the bam file by cluster. But for now, I have this work for me.
You can find the workflow at my github.