 My caption 😄
My caption 😄
An Integrated Platform for Genome-wide Mapping of Chromatin States Using High-throughput ChIP-sequencing in Tumor Tissues
Abstract
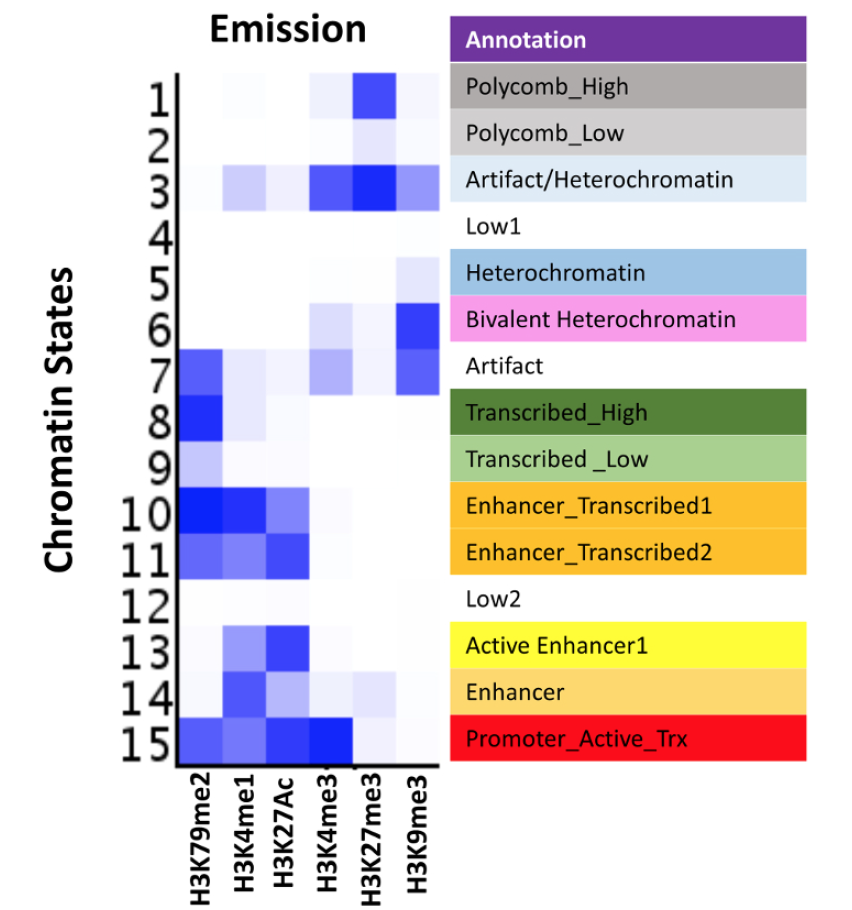
Histone modifications constitute a major component of the epigenome and play important regulatory roles in determining the transcriptional status of associated loci. In addition, the presence of specific modifications has been used to determine the position and identity non-coding functional elements such as enhancers. In recent years, chromatin immunoprecipitation followed by next generation sequencing (ChIP-seq) has become a powerful tool in determining the genome-wide profiles of individual histone modifications. However, it has become increasingly clear that the combinatorial patterns of chromatin modifications, referred to as Chromatin States, determine the identity and nature of the associated genomic locus. Therefore, workflows consisting of robust high-throughput (HT) methodologies for profiling a number of histone modification marks, as well as computational analyses pipelines capable of handling myriads of ChIP-Seq profiling datasets, are needed for comprehensive determination of epigenomic states in large number of samples. The HT-ChIP-Seq workflow presented here consists of two modules: 1) an experimental protocol for profiling several histone modifications from small amounts of tumor samples and cell lines in a 96-well format; and 2) a computational data analysis pipeline that combines existing tools to compute both individual mark occupancy and combinatorial chromatin state patterns. Together, these two modules facilitate easy processing of hundreds of ChIP-Seq samples in a fast and efficient manner. The workflow presented here is used to derive chromatin state patterns from 6 histone mark profiles in melanoma tumors and cell lines. Overall, we present a comprehensive ChIP-seq workflow that can be applied to dozens of human tumor samples and cancer cell lines to determine epigenomic aberrations in various malignancies.
More detail can easily be written here using Markdown and $\rm \LaTeX$ math code.