 My caption 😄
My caption 😄
Evaluating single-cell cluster stability using the Jaccard similarity index
Abstract
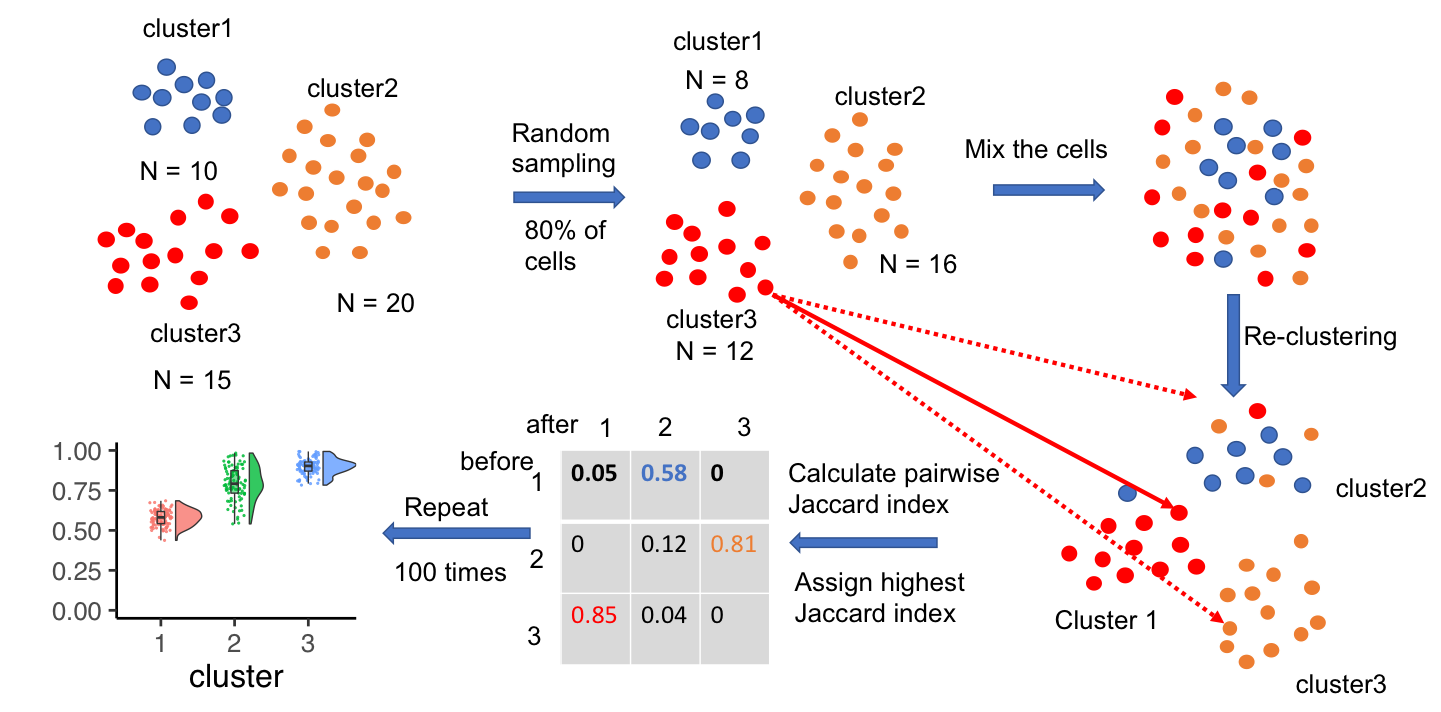
Motivation: One major goal of single-cell RNA sequencing (scRNAseq) experiments is to identify novel cell types. With increasingly large scRNAseq datasets, unsupervised clustering methods can now produce detailed catalogues of transcriptionally distinct groups of cells in a sample. However, the interpretation of these clusters is challenging for both technical and biological reasons. Popular clustering algorithms are sensitive to parameter choices, and can produce different clustering solutions with even small changes in the number of principal components used, the k nearest neighbor, and the resolution parameters, among others. Results: Here, we present a set of tools to evaluate cluster stability by subsampling, which can guide parameter choice and aid in biological interpretation. The R package scclusteval and the accompanying Snakemake workflow implement all steps of the pipeline: subsampling the cells, repeating the clustering with Seurat, and estimation of cluster stability using the Jaccard similarity index. The Snakemake workflow takes advantage of high-performance computing clusters and dispatches jobs in parallel to available CPUs to speed up the analysis. The scclusteval package provides functions to facilitate the analysis of the output, including a series of rich visualizations. Availability: R package scclusteval: https://github.com/crazyhottommy/scclusteval Snakemake workflow: https://github.com/crazyhottommy/pyflow_seuratv3_parameter.
More detail can easily be written here using Markdown and $\rm \LaTeX$ math code.